搜索






 Time:Nov 28, 2024
Time:Nov 28, 2024
1. Research Background
The 2024 Nobel Prize in Chemistry was awarded to DeepMind, as the AlphaFold2 model developed by its team resolved a half-century-old challenge in biology: predicting three-dimensional structures from protein sequences. In the post-AlphaFold era, what constitutes the critical issue in protein science? Undoubtedly, it is protein function. Only proteins possessing desirable functional properties—high activity, high selectivity, and high stability—can become commercially viable protein products. However, predicting protein function remains exceptionally challenging. A fundamental principle holds: altering just 1% of a protein sequence can reduce the activity of the resulting new protein by 95% or even eliminate its biological function entirely. Yet, when AlphaFold2 predicts the structure of these altered sequences, the predicted structures remain largely unchanged. This demonstrates that protein structure does not equate to function; structure is a necessary but far from sufficient condition for function, and a highly inadequate one at that. To address the challenge of protein function prediction, A collaborative team led by Professor Liang Hong at Shanghai Jiao Tong University (comprising the University's Institute of Natural Sciences, School of Physics and Astronomy, School of Pharmacy, Zhangjiang Advanced Research Institute, School of Life Science and Technology, Shanghai AI Laboratory, East China University of Science and Technology's School of Information and Science Engineering, and ShanghaiTech University's School of Life Science and Technology) has dedicated several years to data collection, cleaning, labelling, and AI model exploration, culminating in the development of the Pro series. Their recent work, ‘A General Temperature-Guided Language Model to Design Proteins of Enhanced Stability and Activity,’ published in Science Advances, exemplifies this endeavour. Wet-lab validation demonstrated that the team's Pro-PRIME model achieved positive rates exceeding 30% for top-45 single-point mutations in zero-shot prediction across five proteins (as illustrated in Figure 1). This represents over tenfold improvement in accuracy compared to traditional high-throughput random screening. These mutations enhanced either catalytic activity, thermal stability, resistance to extreme pH conditions, or the ability to synthesise non-natural substrates, demonstrating the model's universal capability. Moreover, through a small-sample fine-tuning approach, highly superior protein mutants emerged within 2–4 rounds of evolution using fewer than 100 wet-lab samples. For instance, T7 RNA polymerase achieved a multi-point mutant with high activity and stability after four rounds of wet-dry iteration. The highest multi-point mutant exhibited a melting temperature (Tm) 12.8°C higher than the wild type, with activity nearly quadrupling that of the wild type. Furthermore, certain product characteristics surpass those of comparable offerings from internationally leading biotechnology companies that have dominated the market for over a decade.
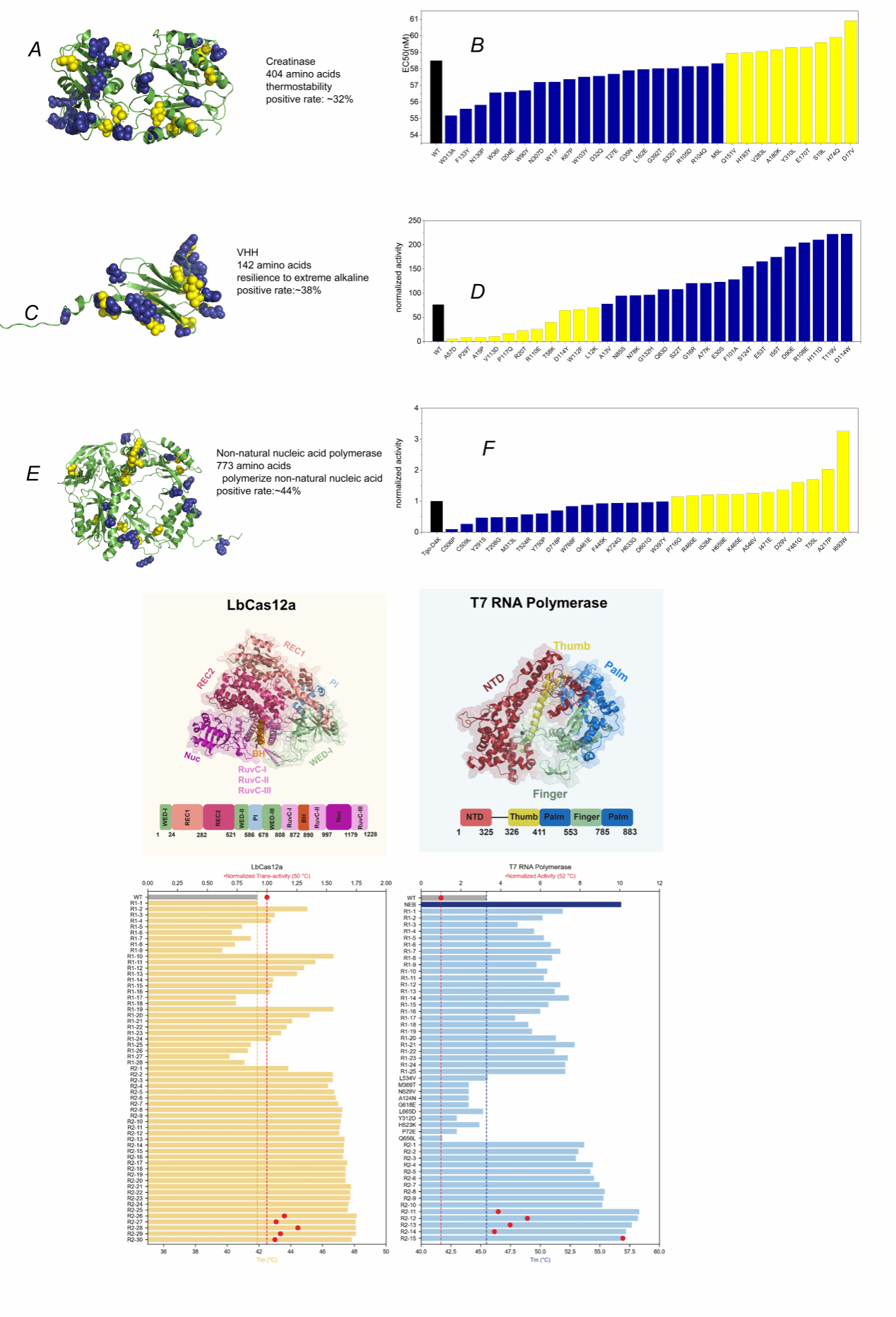
Figure 1: Wet-lab results of Pro-PRIME on five proteins. The top three proteins underwent single-point mutations, while the bottom two proteins—Cas12a and T7 RNA polymerase—achieved 10–15 point mutations within no more than four rounds of dry-wet iterations.
2. Research Methodology
To address these challenges, a research consortium from Shanghai Jiao Tong University, the Shanghai Artificial Intelligence Laboratory, and multiple institutions developed Pro-PRIME (Protein language model for Intelligent Masked pretraining and Environment prediction). This novel deep learning model predicts performance enhancements in specific protein mutants without relying on prior experimental data. Pro-PRIME is trained on a ‘temperature-aware’ language model, utilising a 96-million-entry protein sequence dataset annotated with temperature labels. It combines token-level masked language modelling (MLM) tasks with sequence-level optimal growth temperature (OGT) prediction objectives. Through multi-task learning, a correlation loss term is introduced to align token- and sequence-level task information, enabling the large model to better capture temperature characteristics within protein sequences. This approach inherently biases PRIME towards assigning higher scores to protein sequences exhibiting high-temperature tolerance, thereby optimising stability and biological activity. The Pro-PRIME model, operating entirely without wet-lab data, first leverages PRIME's zero-shot prediction capability to test a small number of single-point mutations. Subsequently, it employs experimental data to iteratively supervise the learning of multi-point mutant predictions. Within no more than four rounds of wet-lab iterations and with experiments conducted on only dozens of mutants, it successfully designed multiple high-performance proteins.
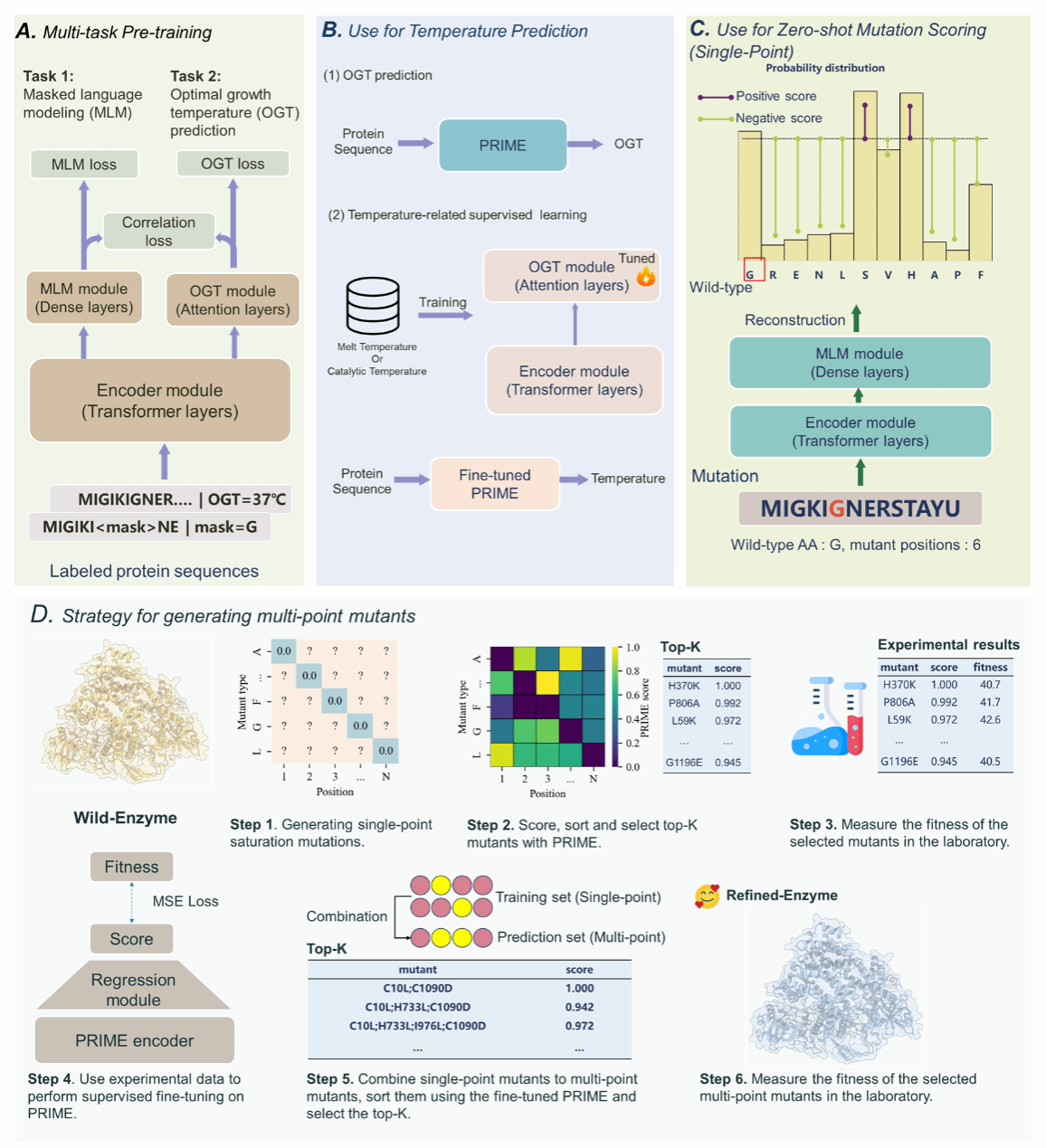
Figure 2: Pro-PRIME's pre-training methodology and single-point mutation zero-shot prediction approach, alongside its dry-wet iterative strategy
3. Research Findings
The PRIME model demonstrates superior predictive capabilities to existing state-of-the-art models across 283 protein experiments within current public mutation databases (ProteinGym and ΔTm); simultaneously achieving superior predictive power over existing state-of-the-art models in both wild-type protein melting temperature (Tm) prediction and optimal enzymatic reaction temperature (Topt) prediction. For wet-lab validation, the team selected five proteins for experimental testing: LbCas12a, T7 RNA polymerase, creatine kinase, synthetic nucleic acid polymerase, and the heavy chain variable region of a specific nanobody. In experimental testing of the top 30–45 single-point mutations, over 30% of AI-recommended single-point mutants demonstrated significant improvement over wild-type proteins in key performance metrics such as thermal stability, enzymatic activity, antigen-antibody binding affinity, non-natural nucleic acid polymerisation capability, or tolerance to extreme alkaline conditions. For certain proteins, the positive rate exceeded 50%. Furthermore, the team demonstrated an efficient method based on PRIME for rapidly generating multi-site mutants with enhanced activity and stability. This streamlined, small-sample refinement approach yielded highly superior protein mutants within 2–4 rounds of evolution using fewer than 100 wet-lab samples. Moreover, in experiments with LbCas12a and T7 RNA polymerase, Pro-PRIME successfully stacked negative single-point mutations to generate positive multi-point mutations. This demonstrates that PRIME learns protein mutation epistatic effects from sequence data, holding significant implications for traditional protein engineering. In summary, Pro-PRIME exhibits broad applicability in protein engineering.
4. Conclusion
PRIME offers a novel approach to protein mutant design that bypasses the need for extensive experimental data accumulation, substantially enhancing the efficiency and accuracy of mutant screening. By effectively reducing reliance on experimental screening, PRIME not only improves success rates in mutant design but also provides innovative solutions to engineering challenges unresolved by conventional methods. Its capability to predict multiple properties of a protein simultaneously furnishes scientists with a valuable tool for successful design even in unfamiliar protein domains.
The potential of this technology extends beyond current research applications, with broad industrial and pharmaceutical implications—particularly in scenarios requiring proteins to exhibit tolerance to extreme temperatures or environmental conditions. Moving forward, this innovation will broaden the application scope of protein engineering, substantially reducing experimental costs and accelerating product development cycles. This research significantly advances the frontiers of protein design, representing a potentially game-changing breakthrough. Concurrently, PRIME's correlation-based multi-task pre-training model offers crucial insights for incorporating biophysical prior knowledge into future large-scale model pre-training.
In summary, PRIME's innovation combines deep learning with big data resources to provide an efficient and practical new pathway for protein engineering. It not only enhances the success rate of protein stability and activity design but also improves experimental efficiency under resource-constrained conditions. With the ongoing development and application of this technology, the field of protein engineering is poised for new breakthroughs, driving vigorous advancement in both scientific research and industrial applications.
Professor Hong Liang from the Institute of Natural Sciences/School of Physics and Astronomy/Zhangjiang Advanced Research Institute, Young Researcher Tan Pan from the Shanghai Artificial Intelligence Laboratory, Liu Jia from ShanghaiTech University, and Song Jie from the Hangzhou Medical College of the Chinese Academy of Sciences are the corresponding authors. Co-first authors include Jiang Fang, PhD candidate from the School of Physics and Astronomy at Shanghai Jiao Tong University; Li Mingchen, intern at the Shanghai Artificial Intelligence Laboratory; Dong Jiajun from ShanghaiTech University; Yu Yuanxi and Wu Banghao from Shanghai Jiao Tong University; and Sun Xinyu from the University of Science and Technology of China. This research received support from the National Natural Science Foundation of China (12104295), the Shanghai Municipal Science and Technology Commission Computational Biology Project (23JS1400600), the Shanghai Jiao Tong University Science and Technology Innovation Fund (21X010200843), the Chongqing Municipal Science and Technology Innovation Major Project (CSTB2022TIAD-STX0017), the Shanghai Artificial Intelligence Laboratory, and the Shanghai Jiao Tong University High-Performance Computing and Student Innovation Centre.
Author: Hong Liang Team
Contributing Unit: Artificial Intelligence Biomedical Centre
Research Centers
Research Capacity
m² Total Area
Building Complexes
沪交ICP备201251287 Copyright © 2025 ZIAS
 Address:No.1308 Keyuan Road, Pudong District, Shanghai
Address:No.1308 Keyuan Road, Pudong District, Shanghai
 Phone:86-21-54740000
Phone:86-21-54740000
 E-mail:zias@sjtu.edu.cn
E-mail:zias@sjtu.edu.cn
