搜索






 Time:Jul 03, 2024
Time:Jul 03, 2024
Recently, Professor Hong Liang's research group from the Institute of Natural Sciences, School of Physics and Astronomy, Zhangjiang Advanced Research Institute, and School of Pharmacy at Shanghai Jiao Tong University, in collaboration with Tan Pan, a young researcher at the Shanghai Artificial Intelligence Laboratory, achieved a significant breakthrough in protein mutation-property prediction. This work employs an innovative training strategy that substantially enhances the mutation-property prediction performance of traditional pre-trained large-scale protein models using minimal wet-lab data. The findings, titled “Enhancing the efficiency of protein language models with minimal wet-lab data through few-shot learning,” were published in Nature Communications, part of the Springer Nature group.
Research Background
Enzyme engineering or protein engineering involves mutating proteins and screening for improved functional products. Traditional wet-lab methods employ greedy algorithm-like search approaches, requiring multiple experimental iterations and repeated validation. These methods demand substantial human resources and time—often taking years to produce a satisfactory protein product—while the mutational sequence libraries they can screen remain extremely limited. Currently, some deep learning methods exist to accelerate this protein mutation modification process. However, achieving high accuracy with these deep learning models requires training on tens of thousands of protein mutation data points. Acquiring this large-scale, high-quality mutation data, in turn, remains a significant barrier for traditional wet lab experiments. Consequently, the industry urgently needs a method capable of accurately predicting protein mutation-function relationships without requiring extensive wet lab data. Existing unsupervised pre-trained models can predict protein mutation-property changes without any wet lab data through zero-shot prediction. However, such predictions often exhibit low accuracy, and these unsupervised models cannot be directly fine-tuned using limited wet lab data.
Research Methodology
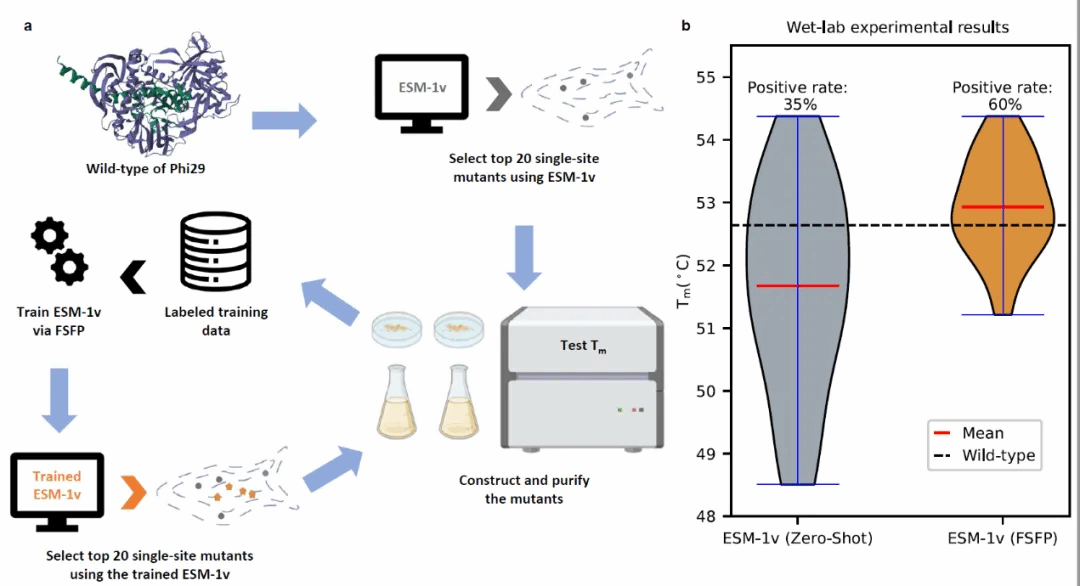
In this study, we propose a solution (FSFP) that integrates meta-learning, ranking learning, and parameter-efficient fine-tuning. This approach enables training protein pre-training models using only a few dozen wet-lab experiments, significantly enhancing the accuracy of protein mutation-property predictions. On the ProteinGym dataset (containing 87 high-throughput mutation datasets), the FSFP method first uses the protein pre-trained model to assess similarity between the target protein and proteins in ProteinGym. It then extracts the two most similar protein datasets from ProteinGym as two auxiliary tasks for meta-learning, while utilizing GEMME scores for the target protein as a third auxiliary task. Finally, the protein pre-training model is trained on a minimal amount of real wet-lab data (tens of samples) using a ranking learning loss function and the Lora training method. Our test results demonstrate that even when the Spearman correlation of mutation-property prediction in the original protein pre-training model is below 0.1, the FSFP method can significantly improve this prediction correlation to above 0.5 by training the model using only 20 arbitrary wet-lab data points.

Research Findings
To investigate the effectiveness of FSFP, we conducted wet-lab validation on a specific Phi29 protein modification case. When trained using only 20 wet-lab data points, FSFP improved the top-20 single-point mutation prediction positive rate of the original protein pre-training model ESM-1v by 25%, and identified nearly 10 entirely novel positive single-point mutations.
Summary
In this work, the authors propose a novel fine-tuning method called FSFP based on protein pre-trained models. By integrating meta-learning, ranking learning, and efficient parameter fine-tuning techniques, FSFP can efficiently train protein pre-trained models using only 20 random wet-lab experiments, significantly improving the model's single-point mutation prediction positive rate. These results demonstrate that the FSFP method holds great significance for addressing the high experimental cycles in current protein engineering and reducing experimental costs.
Professor Liang Hong from the Institute of Natural Sciences, School of Physics and Astronomy, Shanghai Jiao Tong University, and the Zhangjiang Advanced Research Institute, along with Tan Pan, a young researcher at the Shanghai Artificial Intelligence Laboratory, are the corresponding authors. Zhou Ziyi, a postdoctoral researcher at the School of Physics and Astronomy, Shanghai Jiao Tong University; Zhang Liang, a master's student; Yu Yuanxi, a doctoral student; and Wu Banghao, a doctoral student from the School of Life Sciences and Technology, are the co-first authors. This research was supported by the National Natural Science Foundation of China (12104295), the Shanghai Municipal Science and Technology Commission Computational Biology Project (23JS1400600), the Shanghai Jiao Tong University Science and Technology Innovation Fund (21X010200843), and the Chongqing Municipal Science and Technology Innovation Major Project (CSTB2022TIAD-STX0017). Support was also provided by the Shanghai Artificial Intelligence Laboratory, the Zhangjiang Advanced Research Institute of Shanghai Jiao Tong University, and the Shanghai Jiao Tong University High-Performance Computing and Student Innovation Center.
Nature Communications paper link:
Authors:
Hong Liang's Team
Contributing Institution:
Center for Artificial Intelligence in Biomedicine
Research Centers
Research Capacity
m² Total Area
Building Complexes
沪交ICP备201251287 Copyright © 2025 ZIAS
 Address:No.1308 Keyuan Road, Pudong District, Shanghai
Address:No.1308 Keyuan Road, Pudong District, Shanghai
 Phone:86-21-54740000
Phone:86-21-54740000
 E-mail:zias@sjtu.edu.cn
E-mail:zias@sjtu.edu.cn
