搜索






 Time:Sep 11, 2024
Time:Sep 11, 2024
Axial-chiral carbonyl compounds are not only prevalent in natural products, bioactive molecules, drugs, and materials, but also serve as advantageous chiral ligands or organic catalysts with broad prospects in asymmetric catalysis. However, synthesizing such structures often requires cumbersome procedures, limiting their further development. Despite significant advances in asymmetric catalytic synthesis of axial-chiral compounds in recent years, the direct synthesis of axial-chiral carbonyl compounds via asymmetric carbonylation reactions remains an area requiring further exploration.
Catalytic carbonylation represents the most direct and efficient method for synthesizing carbonyl compounds. Since its first realization in the 1830s, carbonylation has become the most industrially applied reaction in homogeneous catalysis, producing over ten million tons of bulk and fine chemicals annually through this technology. In contrast, the development of asymmetric carbonylation has lagged behind, primarily due to the following challenges: (1) High-pressure CO competes with chiral ligands for coordination, weakening the ligands' ability to control chirality; (2) The conversion energy barrier for acyl metal species is high, often constituting the rate-limiting step requiring elevated temperatures; (3) The resulting chiral carbonyl compounds readily racemize within the reaction system. Nevertheless, numerous asymmetric carbonylation reactions have been developed over the past decades. However, the substrate scope remains largely confined to activated alkenes such as norbornene, styrene, and cyclopropene. These reactions are primarily employed for constructing centrally chiral carbonyl compounds, while methods for synthesizing axially chiral carbonyl compounds via asymmetric carbonylation remain to be developed.
Recently, the research group led by Liu Jiawang, Associate Professor on the Tenure Track at the Center for Frontier Science in Transformative Molecules, Shanghai Jiao Tong University, and a resident scientist at the Zhangjiang Institute for Advanced Studies, utilized a chiral palladium catalyst to develop an asymmetric amine carbonylation reaction between racemic bis(aryl)trifluoromethanesulfonate and CO or amine nucleophiles via a substrate dynamic kinetic asymmetric transformation strategy. This approach enabled the efficient and highly enantioselective synthesis of axially chiral amides. Detailed mechanistic studies reveal that intramolecular hydrogen bonding between the heterocyclic nitrogen atom of the amide product and the NH structure accelerates axial rotation, significantly reducing enantiomeric selectivity. The use of cesium carbonate disrupts this intramolecular hydrogen bond, proving crucial for achieving high enantiomeric selectivity. Furthermore, some synthesized amide products can be directly applied as chiral tridentate ligands in copper-catalyzed asymmetric radical reactions, demonstrating their potential utility as chiral ligands.
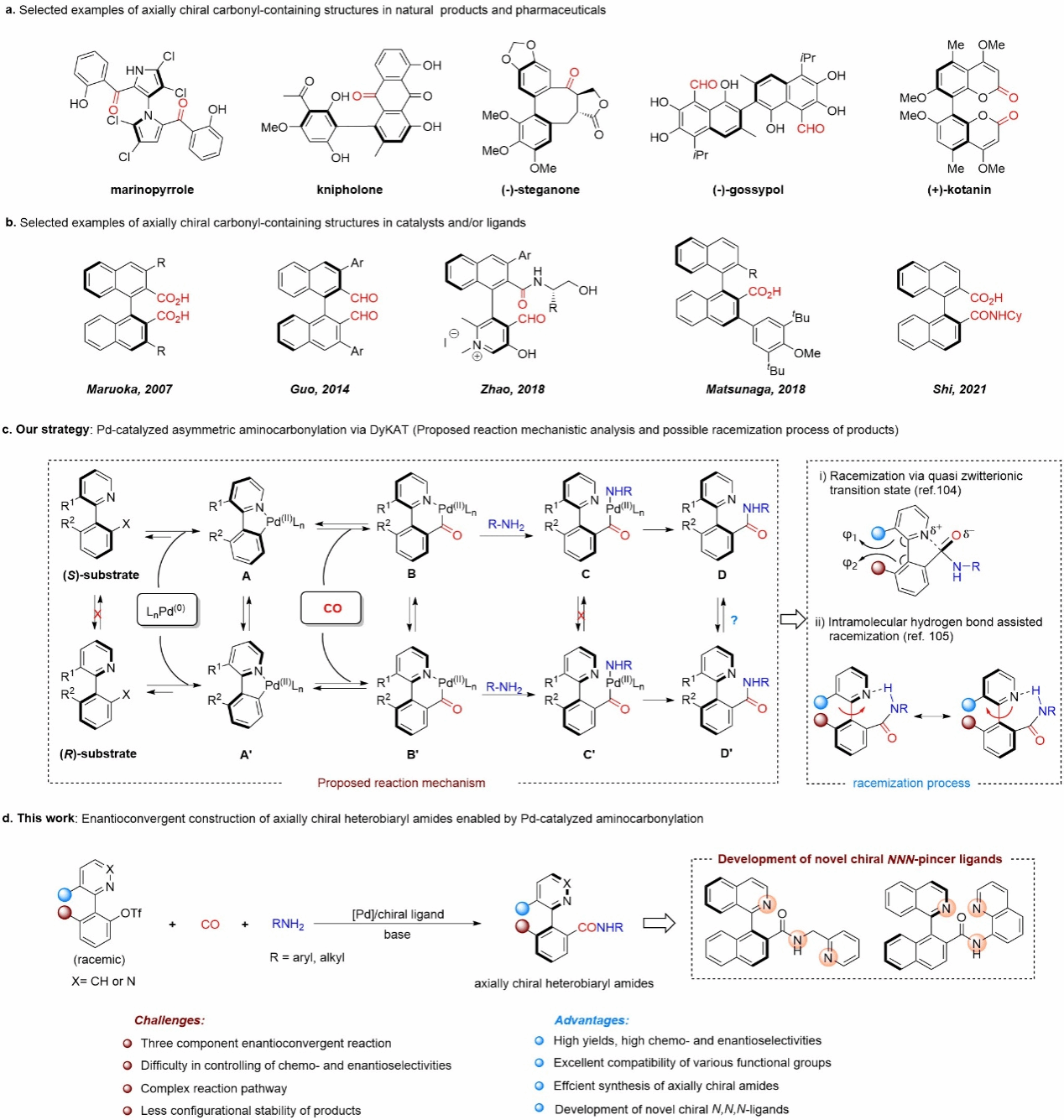
Figure 1 Applications and Reaction Design of Axial-Chiral Carbonyl Compounds
Using racemic aryl trifluoromethanesulfonate compounds 1a and p-toluenamine 2a as template substrates, Pd(OAc)₂ as catalyst, (S,S)-BenzP (L1) as chiral ligand, C₂H₅COCS₂ as the base, and dichloromethane and 1,2-dichloroethane as reaction solvents. Under a CO₂ atmosphere at 10 atm and 80°C for 18 hours, the axial-chiral amide product was obtained in 87% isolated yield with 94% ee.
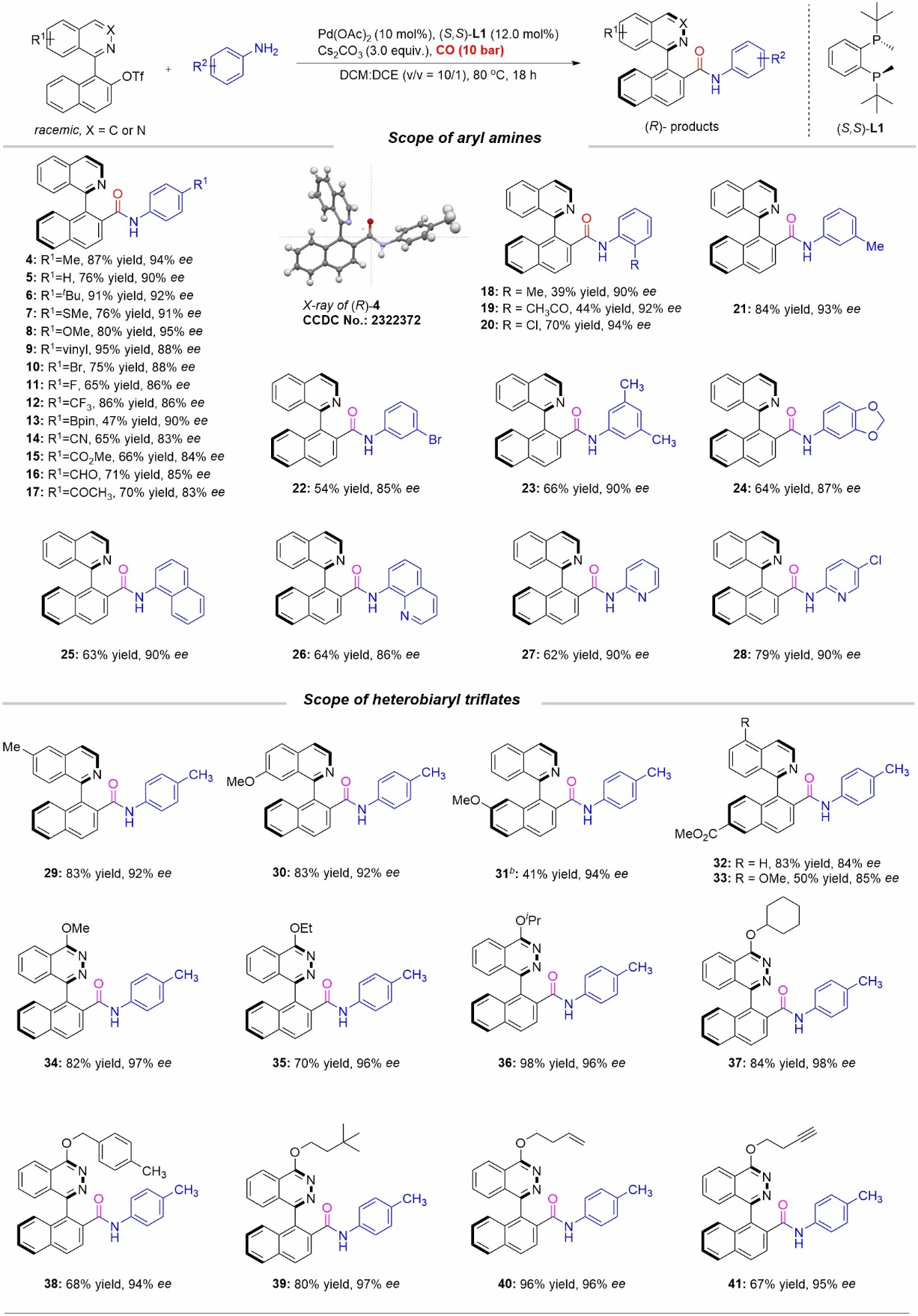
Figure 2 Scope of Aromatic Amines and Bifunctional Aryl Trifluoromethanesulfonate Substrates
Under optimized reaction conditions, the authors first investigated the applicability of aromatic amine substrates (Figure 2). A series of aromatic amines bearing electron-deficient and electron-rich substituents on the benzene ring yielded axial-chiral amides via an efficient kinetic asymmetric amine carbonylation process. Subsequently, the authors investigated diaryl trifluoromethanesulfonate substrates (Figure 2). The reaction retained good universality, yielding chiral amide products in good to excellent yields with outstanding enantioselectivity.
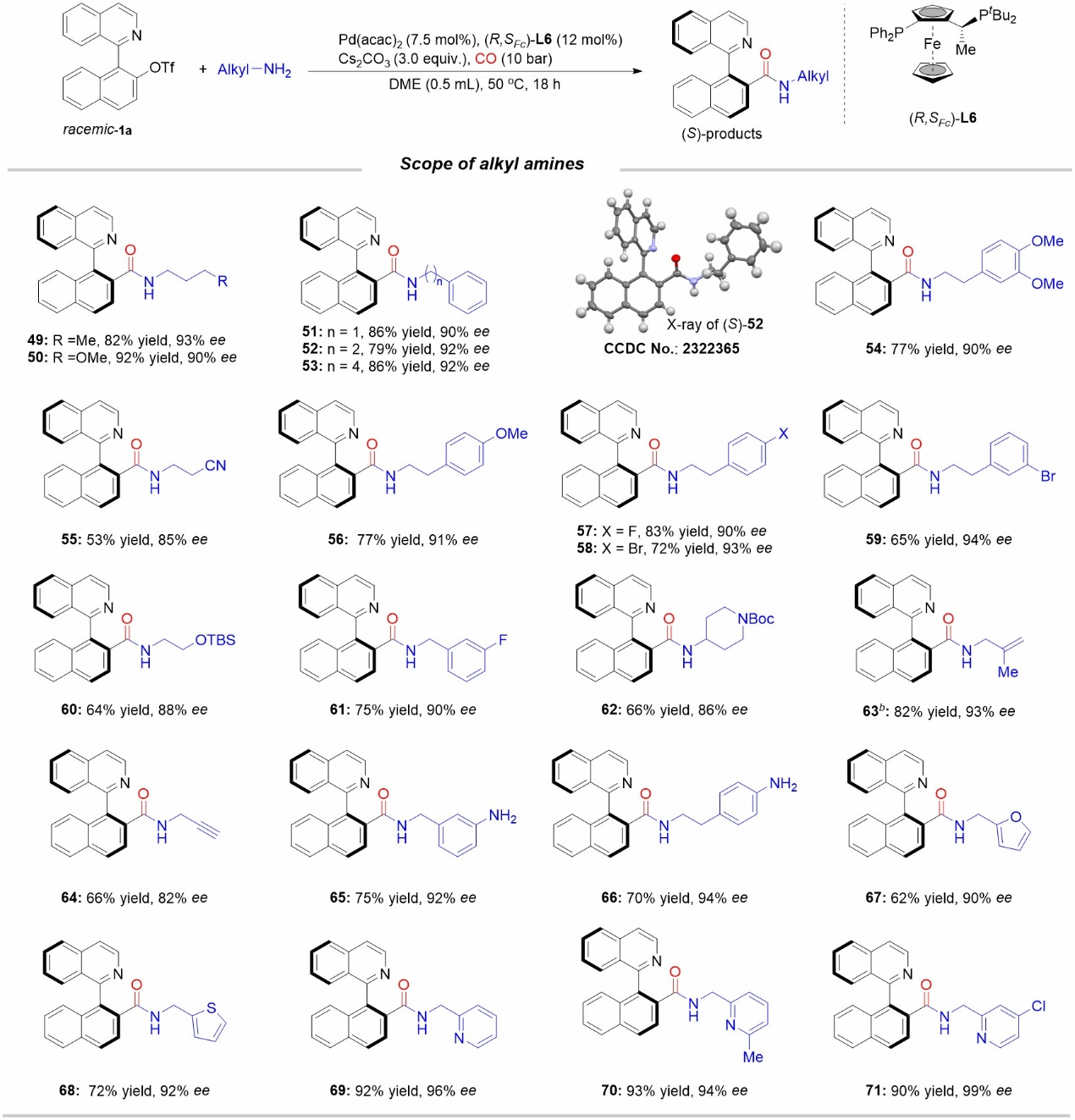
Figure 3 Scope of Alkylamine Substrates
The authors subsequently investigated the use of more nucleophilic alkylamine compounds as nucleophiles. Alkylamines may accelerate the attack on acyl palladium species, potentially adversely affecting the enantioselectivity of the reaction. By optimizing conditions such as the chiral ligand, the authors established a set of asymmetric carbonylation reaction conditions suitable for alkylamines. They systematically investigated the applicability of various substrates, finding good compatibility across primary alkylamines and yielding the corresponding achiral amides.
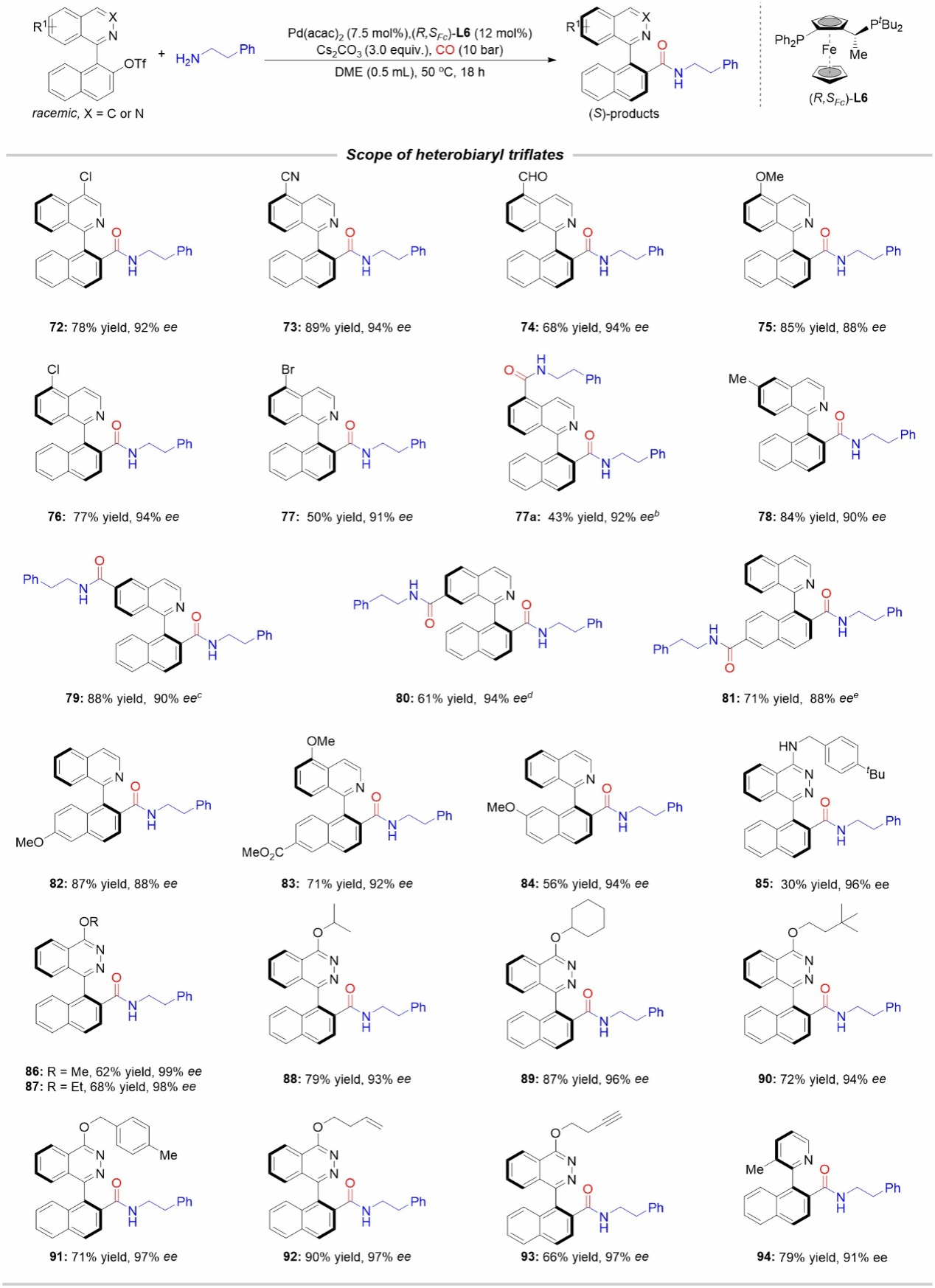
Figure 4: Scope of Substrates for Coupling with Aryl Trifluoromethanesulfonate Esters
The authors subsequently investigated the applicability range of diaryl trifluoromethanesulfonate substrates using 2-phenylethylamine as the coupling reagent. As shown in Figure 4, substrates bearing Me, Cl, CHO, CN, and OMe substituents at positions 4–6 of the isoquinoline ring exhibited good tolerance. Notably, when bromine substituents were present on either the isoquinoline or naphthalene ring, a double amine carbonylation reaction occurred. Other substituents and functional groups were compatible with this system, yielding the corresponding chiral amide products.
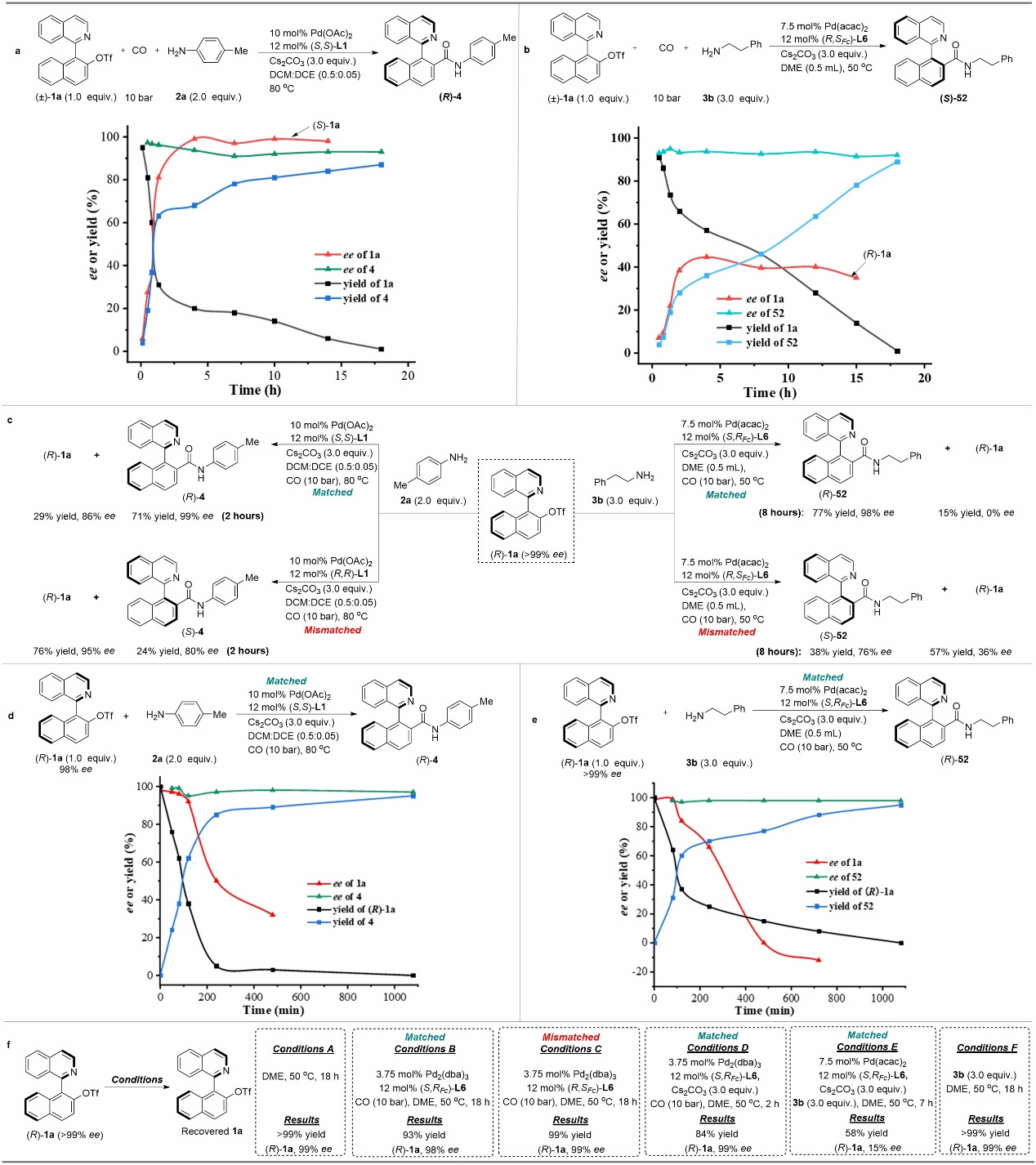
Figure 5 Mechanistic Study
To gain deeper insights into the dynamic kinetic asymmetric amination reaction process, the authors conducted detailed investigations through kinetic experiments and control studies. These examined the interconversion of different enantiomeric substrates involved in the reaction and the recognition of substrates by chiral catalysts. The experiments demonstrated that under standard reaction conditions, substrates with different configurations can undergo reversible interconversion via an oxidative addition-epimerization-reductive elimination pathway.
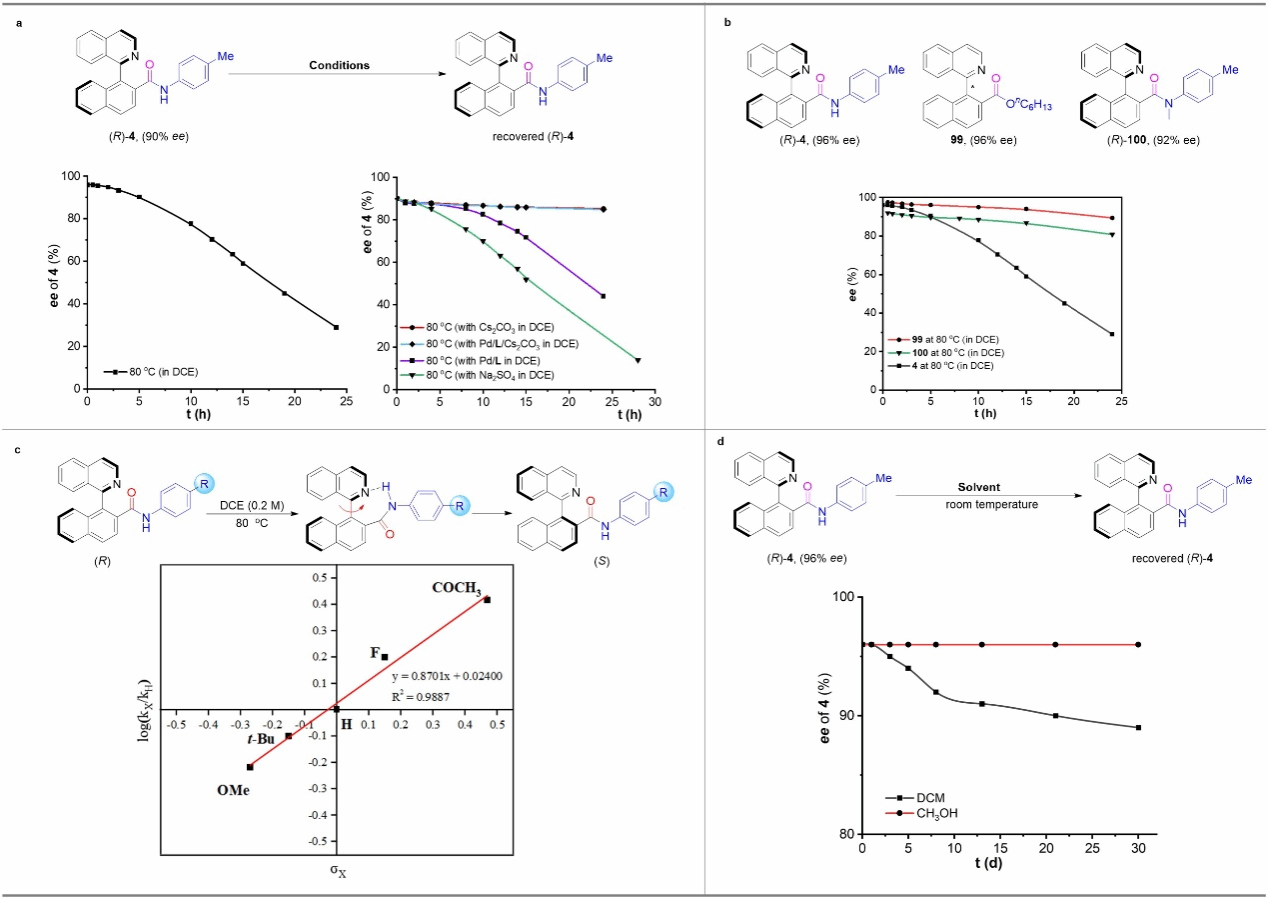
Figure 6 Study on the Stability of the Chiral Axis
During the determination of the rotational energy barrier for the axial chiral amide (R)-4, the authors observed that the enantioselectivity of (R)-4 rapidly decreased from 96% to 29% within 24 hours, contradicting the high ee values achieved in the catalytic reaction. To address this, kinetic sampling experiments were conducted by adding different reactants, revealing that the base plays a crucial role in stabilizing the configuration of product 4. To further investigate how the base inhibits the racemization of product 4, comparative experiments were conducted. The same kinetic sampling experiment was performed on the structurally similar axial-chiral ester 99. Compared to amide 4, ester 99 exhibited a slower racemization rate. Given the stronger electrophilicity of the carbon atom in the ester group, the previously reported racemization pathway via an amphoteric ion transition state was ruled out. Additionally, the authors observed a significant slowing of racemization when using the methylated amide compound (R)-100, suggesting that the NH group of amide products may be associated with racemization. Based on literature reports, the authors speculate that intramolecular hydrogen bonding between the amide NH group and the nitrogen atom of the isoquinoline ring may accelerate axial rotation, leading to product racemization. To obtain further evidence, the authors combined Hammett curves to investigate the effect of aromatic ring substituents on the racemization rate. Experimental results indicate that axial-chiral amides bearing electron-withdrawing substituents on the aromatic ring exhibit faster racemization rates. This is attributed to stronger hydrogen bonding between the NH group of the electron-withdrawing aromatic amine and the nitrogen atom of the isoquinoline ring. Furthermore, the authors stored (R)-4 (96% ee) in methanol or dichloromethane solutions at room temperature for 30 days. They observed a decrease in the product's ee to 89% in dichloromethane, while no reduction in ee was noted in methanol. This suggests that intramolecular hydrogen bonds are likely disrupted by the protic solvent methanol. Based on these experimental results and literature reports, the authors propose that intramolecular hydrogen bonding accelerates the chiral axis rotation of the amide product, while the addition of a base disrupts intramolecular hydrogen bond formation, thereby inhibiting the racemization process of the product. This mechanism is key to achieving high enantioselectivity in the reaction.
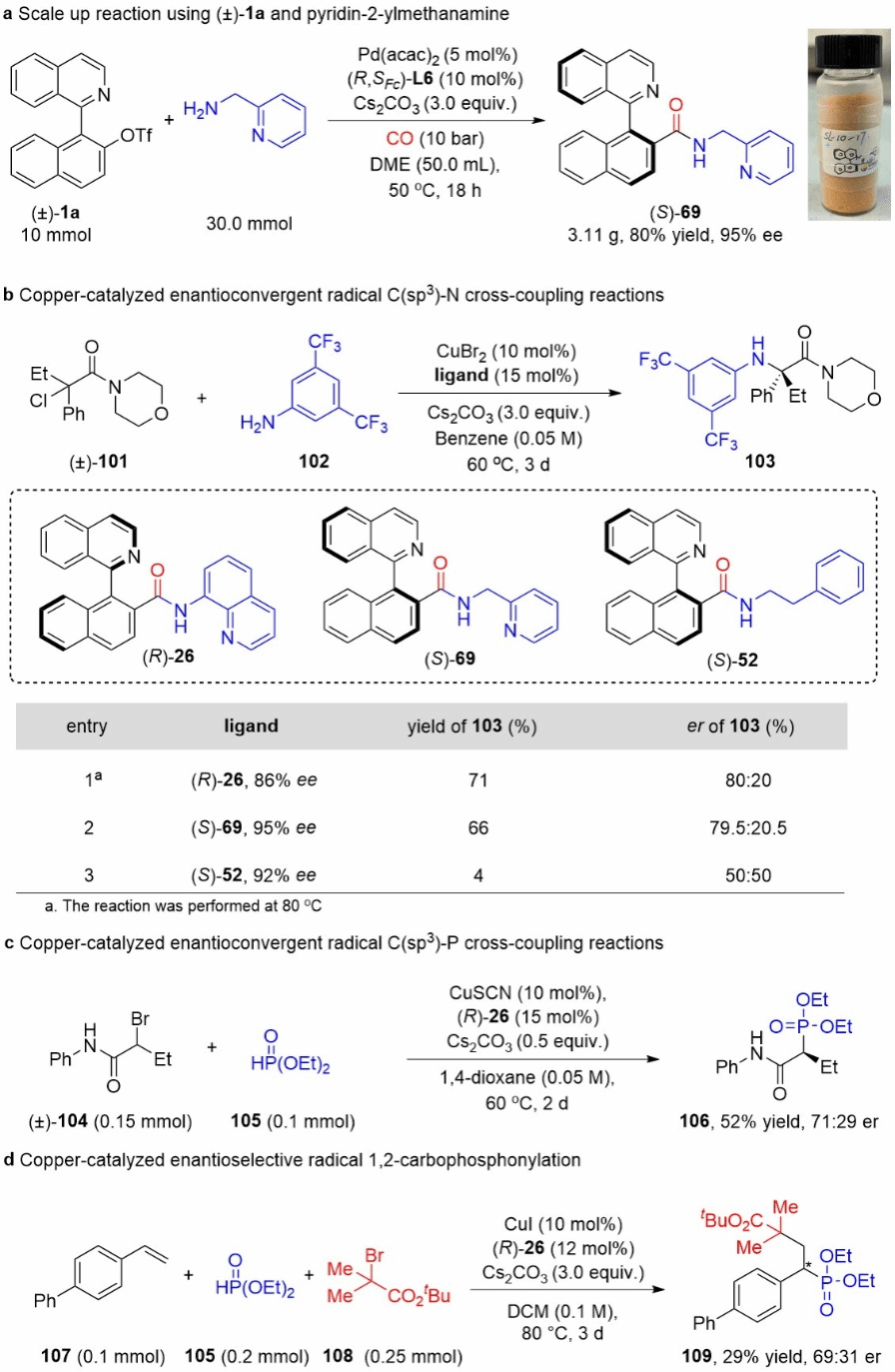
Figure 7 Synthetic Application
Finally, the authors scaled up the reaction system and obtained the corresponding product (S)-69 with equally excellent yield and selectivity. This product can serve directly as a chiral pincer NNN ligand without further modification (Figure 7a). Preliminary results indicate that this class of compounds exhibits certain chiral-inducing capabilities in several copper-catalyzed radical asymmetric reactions, demonstrating their potential as chiral ligands.
In summary, the Liu Jiawang group developed an efficient palladium-catalyzed kinetic asymmetric amine carbonylation system, achieving CO-participating axial-chiral amide synthesis with high enantioselectivity, broad substrate scope, and excellent functional group tolerance. Mechanistic studies reveal that intramolecular hydrogen bonding in the amide product leads to racemization, while the addition of a base disrupts these intramolecular hydrogen bonds—a key mechanism for achieving high enantioselectivity. This reaction not only provides a concise route for synthesizing axially chiral amides but also offers a novel approach for developing asymmetric carbonylation reactions and synthesizing chiral tridentate ligands.
This research was recently published in Nature Communications under the title “Enantioconvergent synthesis of axially chiral amides enabled by Pd-catalyzed dynamic kinetic asymmetric aminocarbonylation.” The corresponding author is Associate Professor Jiawang Liu from Shanghai Jiao Tong University, and the first author is Lei Su, a doctoral candidate at the same institution. This work was supported by the Key Research and Development Program of China, the National Natural Science Foundation of China, the Basic Research Fund for Central Universities, and the Shanghai Rising Star Program.
Article link: https://www.nature.com/articles/s41467-024-51717-8
Author:
Liu Jiawang Team
Research Centers
Research Capacity
m² Total Area
Building Complexes
沪交ICP备201251287 Copyright © 2025 ZIAS
 Address:No.1308 Keyuan Road, Pudong District, Shanghai
Address:No.1308 Keyuan Road, Pudong District, Shanghai
 Phone:86-21-54740000
Phone:86-21-54740000
 E-mail:zias@sjtu.edu.cn
E-mail:zias@sjtu.edu.cn
